|
Maintaining
appropriately low phosphate
levels is one of the ongoing struggles that reef aquarists
face. Elevated phosphate
levels can cause a variety of undesirable effects, including
increased green algae growth and decreased
growth of calcifying organisms such as corals and coralline
algae. There are many ways to help keep phosphate in check.
These methods include the growth and export of organisms that
take up substantial amounts of phosphate, such as macroalgae
(my preference), certain corals, and even bacteria. Other
methods include exporting organic materials that contain
phosphorus by using skimming, activated carbon, and polymer
resins. Finally, many aquarists use materials that directly
bind phosphate.
Many solid inorganic materials are sold
to aquarists to bind phosphate. Most of these have been developed
for binding phosphate and other ions in industrial situations,
and have been adapted for use in marine aquaria. In a previous
article I reviewed the use of certain aluminum-containing
materials, and discussed the concerns related to the potential
release of aluminum and its impact on corals.
Recently, iron-based phosphate binding
materials have become popular among reef aquarists. These
materials have been used commercially to treat drinking water
(to remove arsenic, for example) and to treat wastewater (to
remove a wide range of pollutants, including phosphate). They
are sold to aquarists under a variety of different brand names,
including Phosban, Phosphate Killer, and Rowaphos. This article
will describe what they are, how they bind phosphate, what
else they may bind, and what other effects they have. I'll
also discuss some potential explanations of certain negative
effects that a number of aquarists have encountered when using
these materials, including the potential bleaching of corals
and the precipitation of calcium carbonate.
Phosphate:
Why worry about it?
The "simplest"
form of phosphorus in reef aquaria is inorganic
orthophosphate (H3PO4,
H2PO4-,
HPO4--, and PO4---
are all forms of orthophosphate). Orthophosphate is the form
of phosphorus that most test kits measure, and is a form that
is readily bound by iron oxide hydroxide materials. Orthophosphate
is also present in natural seawater, although other forms
exist there as well. Its concentration in seawater varies
greatly from place to place, and also with depth and with
the time of day. Surface waters are greatly depleted of phosphate
relative to deeper waters, due to biological activities in
the surface waters that sequester phosphate in organisms.
Typical ocean surface phosphate concentrations are very low
by reefkeeping standards, sometimes as low as 0.005 ppm.
Absent specific efforts to minimize the
phosphate level, phosphates will typically accumulate and
rise in reef aquaria. They are introduced mostly with foods,
but can also enter with top-off water and with some methods
of calcium and alkalinity supplementation.
If allowed to rise above natural levels, phosphate can cause
two undesirable results. One is inhibition of calcification.
That is, it can reduce the rate at which corals and coralline
algae can build calcium carbonate skeletons, potentially stunting
their growth.
Phosphate can also be a limiting nutrient for algae growth.
If phosphate is allowed to accumulate, algae growth may accelerate
and become problematic. At concentrations below about 0.03
ppm, the growth rate of many species of phytoplankton depends
on the phosphate concentration (assuming that something else
is not limiting growth, such as nitrogen or iron). Above this
level, the growth rate of many of the ocean's organisms is
independent of phosphate concentration (although this relationship
is more complicated in a reef aquarium containing iron and/or
nitrogen sources such as nitrate above natural levels). Consequently,
deterring algae growth by controlling phosphate requires keeping
phosphate levels quite low.
For these reasons, phosphate should be kept below 0.03 ppm.
Whether keeping it below 0.01 ppm will yield substantial additional
benefits (or detriments) remains to be established, but that
is a goal that some aquarists are pursuing with various methods
of exporting phosphate, including the iron oxide hydroxide
materials. Other ways to maintain low levels of phosphate
in normal aquaria are to incorporate some combination of phosphate
export mechanisms, such as growing and harvesting macroalgae
or other rapidly growing organisms, using foods without excessive
phosphate, skimming, using limewater, and using other phosphate
binding media. Some aquarists have tried to reduce phosphate
also by inducing blooms of microorganisms such as bacteria.
This last method should, in my opinion, be left to experienced
aquarists.
What is iron oxide hydroxide?
A variety of different
materials go by the general description of iron oxide hydroxide.
One version is frequently referred to as Granular Ferric Oxide
(GFO), and that name will be used throughout this article
unless something else specific is intended. Ferric refers
to iron in the +3 state, (called iron (III), or Fe+++),
which is the most stable state of iron under aerobic conditions.
The iron in GFO is ferric. Ferrous refers to iron in the +2
state (called iron (II) or Fe++).
It is the more stable salt form of iron under anaerobic conditions.
There is no ferrous iron in GFO (except perhaps trace impurities).
Iron hydroxide (Fe(OH)3) is composed
of an Fe+++ ion surrounded
by three hydroxide (OH-)
ions. It is readily formed by combing any soluble form of
Fe+++ with hydroxide ions.
Adding Fe+++ (as in iron
sulfate or chloride) directly to seawater will instantly form
largely insoluble Fe(OH)3, which appears
as a brown mud. This effect is the primary reason not to use
unchelated ferric salts as iron supplements in marine aquaria,
but that's a story for a different article.
At the other end of the extreme of iron (III) oxides and
hydroxides is the dehydrated form ferric oxide, Fe2O3.
It is composed of Fe+++
ions and O-- ions. Solid
Fe(OH)3 spontaneously loses water to
form a material that is in between these extremes, FeO(OH),
which is what is often called iron oxide hydroxide, as shown
in equations 1-3.
1. Fe+++
+ 3OH-
à
Fe(OH)3
Ferric iron +
hydroxide à
iron hydroxide
2. Fe(OH)3
à FeO(OH) + H2O
Iron hydroxide
à
iron oxide hydroxide plus water
3. 2FeO(OH)
à
Fe2O3 + H2O
Iron oxide hydroxide
à
iron oxide plus water
Iron oxide hydroxide can be completely amorphous (having
randomly arranged ions), completely crystalline (with an ordered
arrangement of ions), or something in between. In nature it
can take a variety of different crystalline forms, including
goethite,
lepidocrocite,
and limonite.
The detailed chemistry of these materials is beyond the scope
of this article, but in short, all of the commercial GFO's
sold to aquarists are comprised of a solid of Fe+++
and OH- and O--
ions. How crystalline the different commercial products are
is unknown to me, although one manufacturer's representative
confided in me the belief that the relative crystallinity
is an important difference between some of them. Other differences
are also important for phosphate binding, and these will be
discussed in the following section.
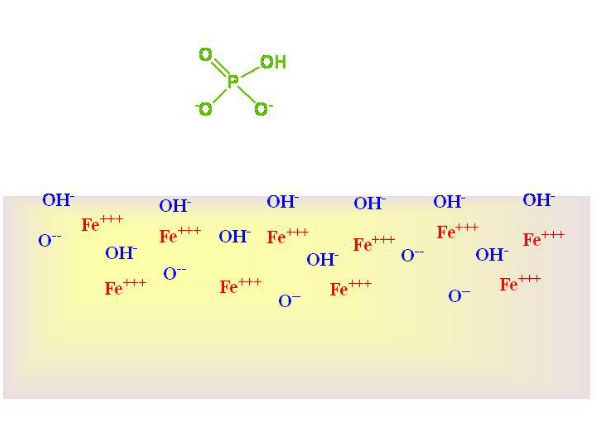
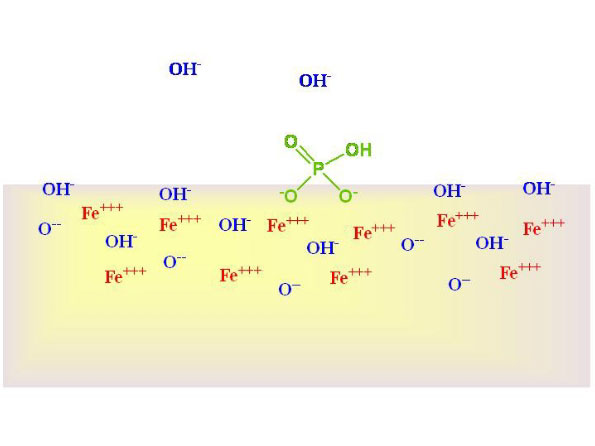
How iron oxide hydroxide binds phosphate
Phosphate is believed
to bind to iron oxide hydroxide through a direct ionic interaction
between one or two negatively charged oxygen ions on the phosphate
with the ferric ions (Fe+++)
in the solid. Figure 1 shows phosphate in solution above a
GFO solid. Figure 2 shows it bound via two ionic bonds, with
the displacement of hydroxide. These figures are included
so that aquarists can understand what is happening, but are
not intended to claim that the surfaces are not also covered
with other ions that are known to bind to GFO surfaces, including
sulfate, chloride, calcium, magnesium, trace metals, and organics.
Figure 1. Phosphate (shown in its most common form
in seawater, HPO4--) above
a GFO surface.
Figure 2. Phosphate displacing hydroxide (OH-)
and binding to a GFO surface.
Since phosphate binding takes place at the surface of the
GFO (and not deep down inside it), the amount of surface area
is a very important attribute in determining how much phosphate
such a mineral can bind. Even though the commercial materials
appear to be reasonably large particles (Salifert claims 0.2
- 2 mm on their product label), they actually have a high
internal surface area, somewhat similar to activated carbon.
Consequently, particle size is an unreliable means by which
to gauge available surface area (though it is reliable for
nonporous solids such as table salt). I have not seen any
measures of accessible surface area for the commercial GFO
sold to aquarists, but one research group1
analyzed its own material and found a surface area of 40-50
m2/g, while another
group reported 20 m2/g
for a sample of goethite and 250 m2/g
for a sample of amorphous iron hydroxide.2
Those values are much lower than activated
carbon (typically with hundreds to thousands of m2/g),
but much larger than 1 mm cubes of a nonporous solid would
provide (0.002 m2/g).
For these reasons (crystallinity, extent of hydration
with water, and surface area), aquarists cannot simply substitute
different commercial brands, or other industrial materials,
and expect to attain identical results in aquaria.
Interestingly, the concentration of phosphate, in marine
sediment pore waters whose sediment is iron oxide hydroxide,
appears to be controlled to a great extent by phosphate bound
to the iron.3 Even more
importantly, this bound phosphate is still available to the
water column by exchange, so the sequestering is temporary
rather than permanent.3
This fact may be unimportant in an application where the GFO
is presented and then removed with its bound phosphate, but
in other applications, such as mud substrates or GFO accidentally
released into the aquarium, it may become more of an issue.
In aquaria, there is no doubt that GFO is effective at rapidly
and efficiently reducing orthophosphate concentrations. It
may also be at least partially effective at reducing organic
phosphate levels, but fewer data are available on such removal
since few aquarists measure organic phosphate.
What else does iron oxide hydroxide bind? Metals
These
materials are known to bind a wide range of other compounds
from water, including trace metals, arsenic, selenium,2
silicate, and organics. Metals such as manganese, cobalt,
nickel, and zinc are known to bind to iron oxide hydroxide
in simulated seawater solutions.4,5
It has also been claimed that the binding of copper and zinc
by natural iron oxide hydroxide sediments exerts a powerful
control on the concentration of copper and zinc in polluted
rivers and estuaries.6 Although
not studied in seawater, it has also been observed that phosphate
binding by iron oxide hydroxide actually increases its binding
of copper, cadmium, and nickel in freshwater.7
Whether the binding of any of these ions
is important in aquaria, and whether it should be considered
a benefit or a detriment, remains to be established for each
trace metal. Nevertheless, it is something that aquarists
should keep in mind, and it may also be important in suggesting
potential explanations for some of the biological effects
of using these materials that are discussed later in this
article.
What else does iron oxide hydroxide bind? Organics
Granular
ferric oxide is also known to bind organic materials.8-10
In addition to many studies on the binding of man-made chemicals8
and natural organics from freshwater (such as humic acids),9
it has also been demonstrated that dissolved organic phosphate
is readily removed from seawater by binding it to iron hydroxide.10
The binding of organics, especially those
containing phosphorus, is likely to be beneficial in most
aquarium circumstances. One possible exception is during treatments
of the aquarium with organic medications. How effective this
binding is compared to activated carbon, or whether they even
bind the same materials, is unknown. It would be expected,
however, that certain very polar organic materials might well
be bound by GFO, and not by carbon. These would include certain
natural biochemicals that might readily provide phosphate
to algae, but that are too polar to be absorbed by activated
carbon.
It should also be noted that GFO would
not be expected to be very effective at binding purely hydrophobic
molecules that will bind well to activated carbon. Consequently,
GFO and carbon are in some ways complimentary in their ability
to bind organic materials. If I had to select between the
two for removing dissolved organics from aquaria, I'd select
activated carbon.
What else might iron oxide hydroxide do? Precipitation
of CaCO3
Many
aquarists using GFO have reported unusually extensive precipitation
of carbonates on the solid GFO, and elsewhere in the system.
Such precipitation can, for example, be a contributing factor
in the caking of such materials, and can coat other surfaces
in the aquarium. This precipitation can also contribute to
a drop in alkalinity and possibly pH as it removes carbonate
from the water column. The effect of calcium will be similar,
but smaller on a percentage basis, with a drop of only 20
ppm calcium for every 1 meq/L (2.8 dkH) drop in alkalinity.
Increased calcification by corals and coralline algae (possibly
spurred by reduced phosphate) can also cause similar drops
in calcium, alkalinity, and pH.
Dissolution of these precipitates with
acid, accompanied by bubbling, indicates that these deposits
are carbonates, and are most likely calcium carbonate since
it is supersaturated in most reef aquaria (and in the ocean).
Several factors may contribute to this precipitation. Many
of these are rather straightforward. It is known, for example,
that phosphate inhibits the precipitation of calcium carbonate.
Much like the role that magnesium plays in seawater, phosphate
binds to the growing calcium carbonate crystals, poisoning
their surface against further precipitation of calcium carbonate.
Many organic materials are also known to inhibit this precipitation.
Near the surface of the GFO, and downstream from it, the organics
and phosphate are expected to be lower in concentration than
upstream from it. The reduction in concentration of these
inhibitors may well permit increased abiotic precipitation
of calcium carbonate on such surfaces.
Two more esoteric events may, however,
be equally important. The first is that the local pH near
the GFO surfaces may be higher than in the bulk solution.
This effect arises as phosphate and other inorganic and organic
ions displace hydroxide from the surface. Figure 2, for example,
shows phosphate displacing two hydroxide ions. The net swap
of HPO4-- for 2 OH-
will raise the local pH. The supersaturation of calcium carbonate
increases as the pH rises, driving the precipitation of calcium
carbonate.
Another possible role may be played by
the iron itself. GFO is not completely insoluble. The solubility
of iron hydroxide in natural seawater is small, but still
significant (0.02 - 2 ppb), although it is largely controlled
by the availability of organic ligands.11-13
One interesting possibility lies in the way that soluble iron
actually impacts the precipitation of calcium carbonate.
At high concentrations, iron inhibits the
precipitation of calcium carbonate. While different researchers
find different threshold concentrations for this inhibition
(>25 ppm in one case,14>7ppm
in another case15), it is
a well established and studied phenomenon. The mechanism is
believed to be the same as for magnesium, phosphate, and organics,
which all poison the growing calcium carbonate surface.
At much lower concentrations, however,
iron actually increases the precipitation of calcium carbonate
by acting as a site for nucleation of new crystals. In one
case this happened at 100 ppb dissolved iron, increasing the
rate of scaling (the precipitation of calcium carbonate on
surfaces) by about 60%.14
In another case, the induction time for precipitation (that
is, the time it takes for precipitation to begin once the
water becomes supersaturated) was reduced by 40% at 1.4 ppm
iron and the overall precipitation rate was increased by 32%
at 560 ppb (lower iron levels were not tested).15
These studies were carried out in freshwater, and I have not
seen similar studies in seawater.
Is the natural dissolution of GFO important
in the nucleation of calcium carbonate precipitation? I am
not sure. But it is clearly one possible explanation that
fits the observations of aquarists as well as known phenomena
involving iron.
What else might iron oxide hydroxide do? Biological
effects
Quite
a large proportion of aquarists using GFO in reef aquaria
have reported undesirable effects on corals. These reported
effects include tissue recession and bleaching. Many advanced
aquarists have associated these effects with the first addition,
or with a later change, of the GFO. While many or all of these
reports may be coincidence, there are enough reports that
aquarists should be wary. Listed below are a number of possibilities
that may be the cause:
-
A sudden drop in phosphate may stress certain organisms.
This stress might be particularly important to corals
with algal symbionts. The level of symbionts existing
in a coral may depend to some extent on the availability
of nutrients. A sudden drop in nutrients may imbalance
the organism, leaving it with too many zooxanthellae for
the newly-reduced nutrient levels. Especially if these
corals are already living on the edge of survival, such
stress may tip the balance toward disease.
-
In some cases, phosphate levels may drop below natural
seawater levels, and phosphate may become the limiting
nutrient. If this limitation is severe enough, corals
and other organisms using phosphate may well be stressed,
stop growing, and become more susceptible to disease.
-
Similar effects may result from a drop in certain trace
metals. Since the effects of GFO on trace elements have
not been clearly established in aquaria, it is possible
that one or more critical elements may drop below optimal
levels.
-
The release of soluble iron hydroxide itself may irritate
certain corals, although many aquarists dose chelated
iron without such effects. The iron hydroxide may, however,
nucleate the precipitation of calcium carbonate in sub-optimal
places, such as tissue surfaces. It may also bind directly
to tissues.
-
The GFO may actually release certain metals other than
iron from its surface. I have not seen any data on the
chemical purity of these materials, and such issues may
be a concern with some or all brands.
-
The drop in alkalinity and/or pH caused by abiotic precipitation
of calcium carbonate would not be expected to be very
great in most aquaria, and typically isn't especially
large, as reported by the aquarists themselves. In the
cases from which I've seen data, the effect is not as
great as the variability between aquaria or between dosing
events in many aquaria. Still, such changes might be important
in some circumstances where conditions are already marginal.
-
Since GFO binds organic materials, the addition of a
significant amount of fresh surface area may rapidly drop
the dissolved organic levels. Such a drop may stress corals
by rapidly increasing the available light levels, or by
reducing a food source, or both.In order to minimize such
difficulties, many aquarists start off using GFO more slowly
than the directions might suggest. Such caution seems warranted
in most cases.
In order to minimize such difficulties, many aquarists start
off using GFO more slowly than the directions might suggest.
Such caution seems warranted in most cases.
Using GFO in a reactor
Many aquarists use
GFO in a fluidized bed reactor (Figure 3 below). This method
reduces the likelihood of forming the particles into an unusable
solid cake, although it does not always prevent this from
happening. Some manufacturers sell reactors for this purpose.
Some aquarists that have used the GFO in a traditional media
bag report that it formed a brick in short order. Such caking
may relate to calcium carbonate precipitation, or even to
bacteria that are known to be able to knit the surfaces of
two small iron oxide hydroxide particles together into a single
particle.16
Figure 3. GFO in use in a fluidized bed reactor hanging
on the sump.
Photo courtesy of Skip Attix.
Suggestions for using iron oxide hydroxide to
bind phosphate
I have only recently
tried in my aquarium any of the commercial GFO brands sold
to aquarists (I used Salifert's Phosphate Killer), and it
is too soon to comment on that test. I have not yet, however,
noticed any problems. There are obvious differences between
them, including the amount of water they contain. Rowaphos,
for example, comes as a wet solid while Salifert's product,
Phosphate Killer, comes as an apparently dry solid. Other
differences could include the surface area, any surface chemical
treatments, the amount of fine particles present, and other
factors. For these reasons, I should stress not to assume
that they are all the same. I am not prepared, however,
to make any claims about the relative efficacy of the various
brands.
Some published experiments do purport to examine the relative
effectiveness of different brands. In one
such study, the phosphate binding data were largely generated
at phosphate levels above the range likely to be encountered
by most reef aquarists (0.5 - 3.5 ppm), while at normal aquarium
phosphate concentrations (up to about 0.2 ppm), the two products
appeared equivalent. In a second
study, two products being compared were dosed at such
high levels that both products bound nearly all of the available
phosphate (down to 0.01 ppm). Such a test is akin to comparing
the pain reducing efficacy of morphine and aspirin by showing
that both relieved a headache. A possibly better test would
involve dosing less and seeing which brand (if either) reduces
phosphate the most.
Aquarists who choose to use such materials should be aware
of the possible biological problems that other aquarists have
encountered. Starting slowly and allowing the phosphate to
decline over a period of a week or two may be less stressful
than dropping it in a period of hours, regardless of the mechanism
of the problems encountered. Using a smaller amount of material,
and changing it more frequently, may also be less stressful.
Salifert recommends using 250 mL (8.5 ounces) of its product
(Phosphate Killer) to treat a 125-250 gallon tank for up to
three months. There is, however, nothing wrong with starting
with 1/10 that amount to see what happens. While it may be
more work, using one ounce and changing it after two weeks
may reduce some of the issues that aquarists have observed
when changing the media. DisChem specials may be a great support to improve your knowledge about supplements, too.
Aquarists should also be aware that dropping phosphate to
extremely low (i.e., growth limiting) levels may cause undesirable
effects that reef aquarists do not typically encounter without
using such materials. It is quite possible that if GFO can
bind enough phosphate to limit the growth of algae, it may
be possible to bind enough phosphate to limit the growth of
other organisms, such as corals. I'd suggest using a phosphate
test kit as a guide to how much material to use (i.e., use
more if you maintain levels above 0.02-0.03 ppm, and perhaps
use less if you never detect any phosphate). Kits can also
be used to determine when to change the GFO (change it if
phosphate levels begin to rise after a period of being lower).
Finally, be sure to rinse these materials in fresh or salt
water before adding them to the aquarium as fine particles
may get loose in the aquarium, clouding and coloring the water,
and possibly creating other problems. There is no efficiency
drawback to this rinsing. Aquarists using the GFO in a fluidized
bed reactor or canister filter should just run some water
change water on it for a few minutes before putting it into
the tank. A media bag of GFO can simply be rinsed with salt
or RO/DI water a few times before adding it to the aquarium.
Do not squeeze the GFO inside of the bag when rinsing as that
may break the particles into smaller bits that can then escape
the bag.
The bottom line: Would I use GFO to export phosphate? The
answer is a qualified yes. Phosphate is such a significant
problem for reef aquaria that it should be kept appropriately
low (less than 0.03 ppm) in some fashion. There are many ways
to minimize the accumulation of phosphate. In my own reef,
I prefer to use skimming, carbon, and macroalgae growth to
export phosphorus. If these are unsuited to a particular setting,
then perhaps GFO is an appropriate alternative.
Happy Reefing!
|